Abstract
GPU and the algorithmic tools are convenient for using, interpreting, and visualizing microbial data. GPU computing has revolutionized the way of performing microbiological research.
In the past, it took a lot of work to sequence the genome of any newly discovered microbe, understand their genomic structures, aligns them, or study the metabolic functioning of microbes. With the advancement of GPU computing, these complex and time-consuming processes have become trouble-free and time-saving.
Conventional computational tools used in this research field have a common feature that limits the functioning and efficiency of these tools by depending on Central Processing Units (CPUs).
To solve this problem, GPUs are earning extraordinary attention from microbiological researchers these days, as GPUs significantly reduce the running time required by CPU-based software and give more detailed views of the biological systems of microbes.
In this review, we will discuss the practical uses of GPU computing in Microbiology.
Rent GPU servers with professional-grade NVIDIA Ampere A100 | RTX A6000 | GFORCE RTX 3090 | GEFORCE RTX 1080Ti cards. Linux and Windows VPS are also available at Seimaxim.
Introduction
GPUs are gaining popularity because they are known for their highly efficient parallel multi-core processors, which provide access to inexpensive and energy-efficient methods.
The nature of the microbial systems under inspection and the purpose of modeling the microbial system both collectively determine the type of GPU-based tools that will be required. So, based on requirements, there are different GPU-based tools available [1].
We use recent GPU-accelerating techniques, which are developed for the alignment of different microbial sequences in predicting the molecular dynamics in microbial cells, searching for microbial structures at both macro and micro levels, and even discovering the pathogen-derived molecule responsible for different diseases in humans [2].
Microbial genome alignment
In microbiology, genome alignment is the most critical step; different strategies for developing less-expensive, high throughput DNA sequences, such as Next Generation Sequencing ( NGS), Illumina sequencing, and Sanger sequencing, have significantly expanded the amount of sequence data that is further used, like genotyping and de novo genome assemblage tasks.
Researchers need faster alignment tools to keep up with this large amount of data. So, GPU computing has provided quicker and high-throughput alignment [3]. Depending on the nature of the alignment, different GPU-based tools are available.
When considering next-generation sequencing (NGS) procedures, which enable the sequencing process by producing a sizable number of subsequences (referred to as “short reads”) of the target genome that must be realigned against a reference sequence (the sequence of our interest),
GPU-powered tools are essential because a typical run for high-throughput NGS approaches generates billions of reads [4].
The alignment problem becomes a complex computational operation that may require much CPU processing time without GPU computing tools. In microbial genome alignment, the genome is not only aligned but also involved in processes like detecting a single nucleotide.
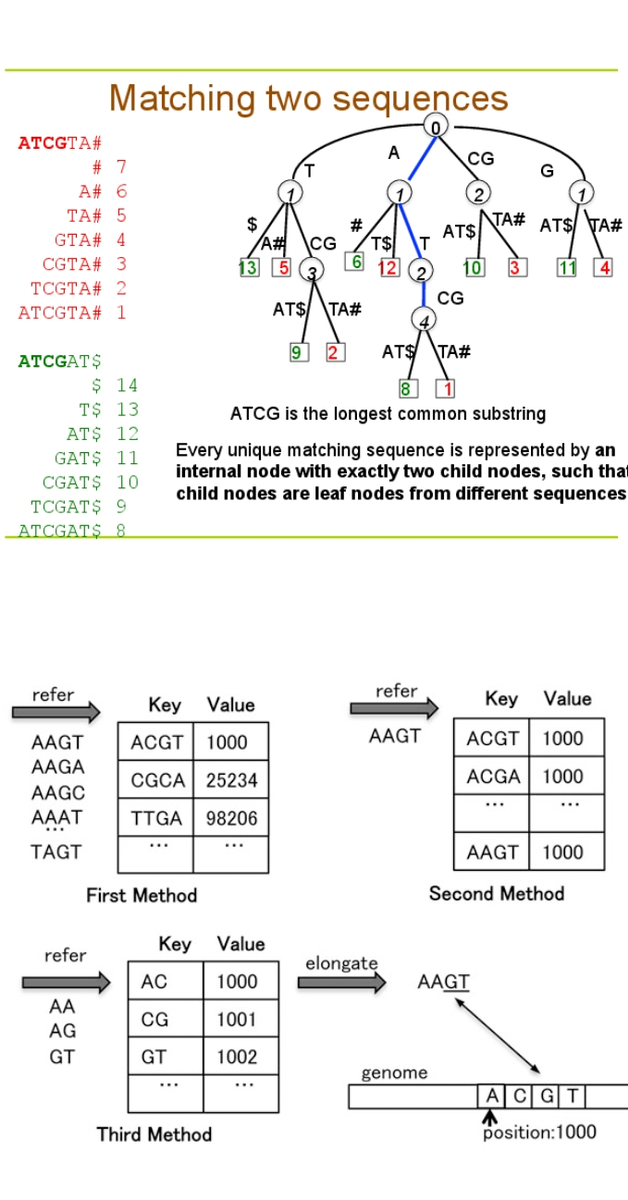
Polymorphism (snips), copy number variations, and reading sequences with gaps. The aligner tools exploit the data in three classes according to the data structure. The data can be hash tables, prefix/suffix trees (shown in Figure 1.1), algorithms, and data structures.
The Burrows-Wheller transform and FM-Index are GPU-based tools that use an algorithm and data structure, while the SARUMAN tool is based on hashing for the alignment processes [5]. Under Burrows-Wheller Transform (BTW), many other GPU-based tools, such as BarraCADA, CUSHAW, and SOAP3, are established on BWT.
SOAP3 (short oligonucleotide alignment tool) is a GPU-based tool that can use both the CPU and GPU to perform heterogeneous computations. SOAP3 was modified to SOAP3-dp, giving foundation to GSNPM(GPU-SNP Mapping), a GPU-based tool to map single nucleotide polymorphisms in the reference sequence. Another exploited version of SOAP3-dp is G-CNV9(GPU-Copy number variation), a GPU-powered tool that checks the operations related to copy number variation detections.
These operations involve the removal of duplicate reads and the filtering low-quality sequences in the reference sequences [6]. Due to GPU acceleration, G-CNV delivers up to 18x acceleration when completing the task. nvBowtie is a GPU-accelerated tool for short-read gap alignment that works at an acceleration speed of 8x.
Later, a MaxSSmap was presented to attain a high level of precision in mapping short reads with gapped alignment. The reassembly of de novo, a target genome from the reads without a reference genome, is complicated.
Two GPU-based software are available for the assembly of de novo sequences: GPU-Euler and MEGAHIT, both used to create a graph of adjacent Sequences [7]. The speed of the GPU-Euler is about 5x.
For the alignment of short reads against a reference genome or to identify similar ones by comparing the query sequence with a library sequence, for this purpose, another GPU-accelerated tool is G-BLSTN (GPU-Basic local alignment search tool) [8]. G-BLATN offers a 14.8x speed-up.
An alternative tool is MuMmerGPU 2.0, which shows data in the form of suffix trees with a speed-up of 4x. GPU computing also solves the problem of sequence similarity during detecting familiar motifs [9].
The GPU-based tool mainly used for this issue is HMMER3, but it cannot provide any speed-up, so CUDAMPF is used as an alternative, which gives a 37x speed-up. GPU computing has also solved the problem of multiple sequence alignment [10].
Two GPU-based tools are used to solve this problem: CUDACLustal, which performs alignment at the speed-up of 30x. While on the other hand, GPU-REMuSic is preferred over CUDACLustal because it distributes the calculations across multiple GPUs [11].
Visualization of microbial molecular dynamics
The molecular visualization tool based on GPU computing was developed almost 20 years ago, allowing researchers to see the structure of microbial proteins and biomolecules and analyze their dynamics. This will enable them to understand better their biological functions and how pharmaceutical compounds interact with these structures. Understanding these complex biomolecular systems helps them do their day-to-day work [12].
A researcher has to perform many computationally demanding calculations to understand how these structures work, and some of these calculations are tremendously demanding.
GPUs have become an effective means for researchers to carry out rapidly and conveniently on a desktop computer, allowing them to do their work much more efficiently [13]. It will enable them to look at much larger and more complicated molecular systems than was possible in the past.
We can now observe microbial molecular surfaces at high magnification, which previously were far too large to be kept in real-time on the CPU, and as you can imagine, if we have to do this on the CPU, which is the host computer machine, may have far more memory than GPU.
Still, it is much slower and unable to maintain an interactive display rate. So, one of the new features in the latest versions of tools used for the visualization and analysis of microbial molecular dynamics is this significantly increased memory [14].
Molecular orbital display algorithm for quantum chemistry datasets and that algorithm makes use of exponentials and reciprocal square roots and other special functions that, when excited on GPU these, when performed on GPU, execute in a handful of clock cycles when executed on the host machine for various reasons [15].
The CPU does not have high-performance hardware for that kind of work, so this is the speed-up we see in the case of the GPU. GPU can perform these calculations quickly; you would have over a hundred CPU cores to do the same work.
Over the years, we have been adapting visualization of microbial molecular dynamics to scale up to much larger size problems. The main goal of work that the researchers have done in this field is to use the computer as a form of computation microscope.
So, the goal of this visualization of microbial dynamics using GPU is to use a computer to see time scales and structural details that are currently inaccessible to experimental imaging methodologies, allows the researchers to view large structures like HIV capsids in the laboratory and enable the molecular surface representation to do animations and things that would not be possible without that feature [16].
For electrostatic interactions of protein systems having 38453 to 1004947 atoms, we primarily use Mashino et al. It has a speed-up of 100x. There is another GPU-based software OpenMM which can deal with systems having 23558 atoms [17].
The area where you get the most extraordinary GPU acceleration are algorithms that have components in them that, unsurprisingly, have some similarity to what GPUs do in their day job, which is the shading of all pixels on the screen, so things that contain transcendental functions tend to be very fast on GPUs; so to give an extreme example, one of the fastest algorithms in the visualization of molecular dynamics is GPU computing [18].
Currently, we can produce 100 million and 200 million atoms simulations. So, for stimulation, researchers combine x-ray crystallography images with their electron density map, and this step needs a lot of time if done without GPU [19].
Microbial molecular docking
The purpose of microbial molecular docking using GPU computing is to recognize the accurate matching between two microbial molecules, which can be a protein with protein, a protein with ligand, or a protein with DNA complex.
This process also involves drug-designing approaches. In this method, the researcher usually observes the structure of molecules, which can be rigid, semi-rigid, or flexible, conformational space, and the binding sites of molecules. Ritchie and Venkaleaman first presented GPU-based protein-protein interaction of the rigid body stage model.
They used spherical polar Fourier protein docking, which relies on the CuFFT library and is mainly GPU-based, with a speed-up of 45x [20].
Simosen et al.present Moldock, a GPU-based tool, to search for drug candidates focused on flexible molecular locking of protein-ligand molecular complexes. This method assists in finding an appropriate binding site to make binding between protein and ligand possible.
This tool accomplishes this task with a speed-up of 27.4x. To perform molecular docking of protein with DNA recently, the researchers are using appAlign and MEGA DoCK. For large-scale protein structure alignment, ppsAlign is the most frequently used tool which uses GPU for structural comparison with a speed-up of 39x [21].
On the other hand, MEGA DOCK uses Katachalski-kalzir, the algorithm that requires supercomputers with GPU for analyzing protein with protein rigid stage interaction. The supercomputer involved in this process is mostly TSUBAME 2.5, equipped with GPU, which can study 3097 protein pairing with 176 receptors and ligands.
Ray casting is the tool that researchers use to visualize the docking of inhibitors or other small molecules with the protein surface having small pocket-like structures and for visualization of activated new molecules in any biochemical assay. Ray casting with an affordable GPU can complete this task with a speed-up of 27x [22].
Prediction and searching of microbial molecular structures
Researchers use GPU computing to recognize stable secondary structures of RNA or single-stranded DNA molecules with minimum free energy [23]. To identify secondary structures of unfolded RNA, researchers use UNAFold, a GPU-based tool with a speedup of 17x.
For the identification of tertiary structures of microbial proteins, researchers use MemHPG, which can create a three-dimensional configuration of the protein of interest by calculating the inter-atomic spaces.
To search for a protein from a protein database based on its three-dimensional structural similarities, researchers use GPU-CASSERT has a speedup of 180x [24].
Mimicry of microbial cells
In microbiology, the physiology and biochemical processes of microbial cells are studied extensively. By using GPU computing, researchers can now efficiently check the physiology of microbial cells by creating a mimic of 3D minimal cells.
The researcher used minimal cells for this process because mini cells are more uncomplicated than the original microbial cells. So they can get readily displayed digitally. Using GPU computing today, we can replicate information from 7000 genes in just 20 mins of cell cycles of the bacterial cell.
We can create a mimic display of 3D minimal cells by using GPU-accelerated software like Lattice Microbes. For this, researchers usually use mycoplasma, the simplest known living cell, and make a model of 500 genes.
GPU-accelerated mimic model of mycoplasma can study minor structural molecules like lipids, amino acids, proteins, Nucleotides, and even membranes.
With the help of Lattice Microbes, researchers can furthermore predict which microbial cells use a large amount of energy to transport molecules out of the membranes if microbial cells under study use a large amount of energy so the cell can be the parasitic line.
This GPU-based tool can also study the growth and cell division process. Researchers use Titan, one of the fastest GPU-based supercomputers, to stimulate the immature stage of retrovirus [25].
Understand the epidemiology of pathogenic microbes
At a large scale, due to numerous genomic data of pathogenic microbes, it is challenging to characterize the mode of transmission of infectious diseases in humans. There are a lot of methods to analyze the raw sequence data of variants, but all these methods are slow and need a lot of struggle. So, GPU computing is an accelerated, reproducible method to analyze the genomic variants of different pathogenic microbes.
We can understand that particular disease’s epidemiology and evolutionary process by analyzing pathogen genomic data with GPU-based tools. Researchers analyze local and global data by monitoring the pathogen’s geographic origins, transmission, and variants.
To meet a large number of whole genome sequencing data of pathogenic variants, researchers are now using Clara Prabricks, GPU-based software that allows them to perform secondary analysis of genomic sequenced data of variants [26].
Parabrick tool uses GPU and CPU hybrid systems to manage and distribute the workflow. It uses a hybrid system of GPU-CPU because all the computational operations need GPU, and to read inputs, calculate graphs, and give the processed information in the form of output requires a CPU. The parabrick tool requires 4 GPUs and 16X CPUs with a speed-up of 27x.
The parabrick is known for dealing with epidemiologic studies associated with P. falciparum, the primary cause of malaria in humans. Parabrick reduces the execution time by simplifying the pathogen genomic mapping and variant recognition and generates output in less than 5mins with 99.9% accuracy.
Still, if we do the same task without GPU, it takes 2 to 3 hrs. Parabrick is not only for malaria analysis; it can be widely used to study the genetic variations and genomic epidemiology of other human pathogens that involve bacteria and other protozoa [27].
Discussion
Despite the numerous benefits of using GPU computing, there are still some issues that researchers have to face while using this technology. The supply of memory is a possible problem for GPUs.
Many applications need much more memory than several gigabytes currently available on top-tier GPUs, especially those that process genome-wide data. CPUs continue to perform significantly better than GPUs when it comes to memory.
For this short memory issue, there is software that gives direct access to the CPU’s memory called pinned memory. For truthfulness, we reveal that, on the most recent implementations, running in parallel kernels can be performed on a single GPU, offering a hybrid SIMD-MIMD operation.
GPUs are primarily designed to provide “Same Instruction Multiple Data” (SIMD) parallelism, in which all cores in the GPU are assumed to operate the same commands on different input data.
This is considerably different from the “Multiple Instruction Multiple Data” (MIMD) techniques used in parallel and grid computing, where each data processing unit is independent, asynchronous, and capable of handling a variety of data formats and running a variety of programs.
The CPU program cannot be instantly converted to the GPU’s framework since the current CPU architectures do not frequently utilize SIMD as an operation mechanism.
The CPU code needs to be updated for GPUs because they have completely different architectures from CPUs, offer a diverse range of operations, and use various libraries. In addition, to better match and fully utilize this architecture, the existing algorithms typically need to be redesigned due to the complicated system of memories and the limited number of high-performance memories accessible on GPUs.
Consequently, from the perspective of a computer programmer, GPU programming is still a challenging task. So, it proves that GPU computing requires a lot of work, and programmers are exploiting GPU computing to make it better for use in the future.
Conclusion
In this review article, we have discussed and mentioned the uses of GPU computing in microbiology. We have discussed all the recent and advanced tools based on GPU computing.
Here, we draw attention to the fact that while the speed-up values given in the literature demonstrate that GPUs are a potent tool for significantly reducing running times, most of the estimated accelerations may be debatable because there may be potential for more CPU code improvement.
Most effective tools require a customized implementation to fully utilize the GPU and its predicted peak performance. The best GPU-powered solutions share the following traits: they take advantage of high-performance memories and work to limit access to global memory by utilizing data structures that have been GPU-optimized.
These characteristics are crucial for practical GPU implementations, optimal memory layouts, and task distribution that is intelligently distributed over multiple threads with less branch divergence. In other words, we caution against naive attempts to convert existing technology because such attempts are typically bound to failure.
Although the speedup obtained with optimized CUDA code is currently significant, it is essential to note that the continuous advancement of the manufacturing process for GPU-enabled graphics cards is anticipated to widen the efficiency gap compared to CPUs.
Because more recent video cards have more cores and more high-performance resources (like registers, shared memory, and cache), which remove the primary barriers preventing full utilization of GPUs in several existing implementations, GPU-powered software generally performs faster when running on these cards.
So, we get highly detailed videos and images of biomolecules of microbes with greater precision.
References
- Joubert W, Archibald R, Berrill M, et al. Accelerated application development: The ORNL Titan experience. Comput Electr Eng 2015;46:123–38.
- He M, Petoukhov S. Mathematics of Bioinformatics: Theory, Methods, and Applications. Hoboken, NJ: John Wiley & Sons, 2011.
- Alberghina L, Westerhoff HV.. Systems Biology: Definitions and Perspectives, Vol. 13 of Topics in Current Genetics. Berlin, Germany: Springer-Verlag, 2005.
- Karr JR, Sanghvi JC, Macklin DN, et al. A whole-cell computational model predicts phenotype from genotype. Cell 2012;150(2):389–401.
- Pop M, Salzberg SL. Bioinformatics challenges of new sequencing technology. Trends Genet 2008;24(3):142–9.
- Intel® SSE4 Programming Reference. Reference Number: D91561-003. Intel Corporation, Denver, CO, USA, 2007.
- Iosup A, Epema D. Grid computing workloads. Internet Comput IEEE 2011;15(2):19–26.
- Foster I, Kesselman C.. The Grid 2: Blueprint for a New Computing Infrastructure. Los Alamitos, CA: Elsevier, 2003.
- Dematté L, Prandi D.. GPU computing for systems biology. Brief Bioinform 2010
- Harvey MJ, De Fabritiis G.. A survey of computational molecular science using graphics processing units. WIREs Comput Mol Sci 2012.
- Klus P, Lam S, Lyberg D, et al. BarraCUDA—a fast short read sequence aligner using graphics processing units. BMC Res Notes 2012;5:27.
- Liu Y, Schmidt B, Maskell DL.. CUSHAW: a CUDA-compatible short read aligner to large genomes based on the Burrows–Wheeler transform. Bioinformatics 2012;28.
- Torres JS, Espert IB, Dominguez AT, et al. Using GPUs for the exact alignment of short-read genetic sequences by means of the Burrows-Wheeler transform. IEEE/ACM Trans Comput Biol Bioinform 2012;9(4).
- Schulz R, Lindner B, Petridis L, et al. Scaling of multimillion-atom biological molecular dynamics simulation on a petascale supercomputer. J Chem Theory Comput 2009;5(10).
- Sauro HM, Harel D, Kwiatkowska M, et al. Challenges for modeling and simulation methods in systems biology In: Perrone L, Wieland F, Liu J, et al. (eds). Proceedings of the 38th Conference on Winter Simulation. New York: IEEE, 2006.
- Klepeis JL, Lindorff-Larsen K, Dror RO, et al. Long-timescale molecular dynamics simulations of protein structure and function. Curr Opin Struct Biol 2009;19(2)
- Susukita R, Ebisuzaki T, Elmegreen BG, et al. Hardware accelerator for molecular dynamics: MDGRAPE-2. Comput Phys Commun 2003;155(2):115–31.
- Liu W, Schmidt B, Voss G, et al. Accelerating molecular dynamics simulations using Graphics Processing Units with CUDA. Comput Phys Commun 2008;179(9).
- Mashimo T, Fukunishi Y, Kamiya N, et al. Molecular dynamics simulations accelerated by GPU for biological macromolecules with a non-Ewald scheme for electrostatic interactions. J Chem Theory Comput 2013;9(12).
- Grosdidier S, Totrov M, Fernández-Recio J.. Computer applications for prediction of protein–protein interactions and rational drug design. Adv Appl Bioinform Chem 2009;2:101
- Ritchie DW, Venkatraman V.. Ultra-fast FFT protein docking on graphics processors, Bioinformatics 2010;26(19).
- Rizk G, Lavenier D.. GPU accelerated RNA folding algorithm In Allen G, Nabrzyski J, Seidel E, et al. (eds). Computational Science–ICCS 2009. 9th International Conference, 2009 Proceedings, Part I, Vol. 5544 of Lecture Notes in Computer Science. Heidelberg, Berlin: Springer, 2009, 1004–13.
- Markham NR, Zuker M.. DINAMelt web server for nucleic acid melting prediction. Nucleic Acids Res 2005.
- https://analyticsindiamag.com/scientists-use-nvidia-gpu-to-mimic-living-cell/2022
- Gardy JL, Loman NJ. Towards a genomics-informed, real-time, global pathogen surveillance system. Nat Rev Genet 2018;19(1).
- DePristo MA, Banks E, Poplin R, et al.. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 2011;43(5)
- MalariaGEN Plasmodium falciparum Community Project. An open dataset of Plasmodium falciparum genome variation in 7,000 worldwide samples. Wellcome Open Research 202.
- https://www.seimaxim.com/dedicated-servers/gpu