GPUs are excellent for speeding up metagenomics analysis. Consider a scenario where researchers could examine the genomes of all the bacteria in a single soil or water sample in real time. This is the capability of graphics processing units or GPUs. Therefore, it enables us to speed up metagenomics analysis.
The study of all the bacteria in a specific environment’s genetic makeup is known as metagenomics. It is an effective method for gaining insight into microbial communities’ variety and operation and discovering novel microorganisms and microbial products. However, metagenomics analysis can be costly computationally, mainly when working with massive datasets. GPUs have a role in these as highly specialized processors with a parallel computing architecture.
Using GPUs, scientists may now analyze metagenomics data considerably more quickly than in the past.
It allows researchers to learn more about the microbiome of humans and bacteria that dwell in harsh conditions.
For instance, scientists are utilizing GPUs to investigate how the gut microbiome interferes with human health and causes disease.
Additionally, they are employing GPUs to find novel microorganisms that might be useful to create novel antibiotics or other treatments.
GPUs are also helpful to research the bacteria that can survive in harsh conditions, like deep-sea hydrothermal vents and hot springs.
For availing GPU servers with professional-grade NVIDIA Ampere A100 | RTX A6000 | GFORCE RTX 3090 | GEFORCE RTX 1080Ti cards. Linux and Windows VPS are also available at Seimaxim.
This research makes it possible to understand how microorganisms adapt to these challenging circumstances and their function in these ecosystems.
What is Metagenomics?
The study of the genetic makeup of every bacterium in a specific environment is known as metagenomics.
It is an effective method for learning about the diversity and purpose of microbial communities as well as for finding novel microorganisms and microbial products.
In a metagenomics analysis, all the bacteria in a sample have their DNA sequenced. After that, computer techniques help identify the microbes and estimate their functions.
The variety of species and behavior of microbial communities can be studied using this information. Moreover, new bacteria and microbial products can be found.
Our understanding of the microbial world has undergone a revolution thanks to metagenomics.
It indicates that contrary to what we previously believed, microorganisms are significantly more diverse and crucial to the ecosystem.
Metagenomics is also helpful in creating new biofuels, other goods, and new diagnostic methods and disease treatments.
How GPUs Accelerate Metagenomics Analysis
Here are some specific examples of how GPUs are used to accelerate metagenomics analysis
GPU-accelerated sequence alignment for
Metagenomics Analysis
GPU-accelerated sequence alignment accelerates the sequence alignment process by exploiting the parallel processing characteristics of GPUs.
The method of comparing two DNA or RNA sequences to find common areas is known as sequence alignment.
It is a crucial step in many bioinformatics applications, including metagenomics analysis, genome assembly, and gene prediction.
Sequence alignment works well with GPUs since they can handle numerous computations simultaneously. It is significant because sequence alignment entails comparing two sequences at every conceivable place.
Because sequence alignment sometimes entails comparing sequences to vast databases of reference genomes. Therefore, GPUs’ ability to access massive datasets swiftly is crucial.
Smith-Waterman algorithm
There are numerous different GPU-accelerated sequence alignment algorithms.
Using a parallel version of the Smith-Waterman algorithm is one such strategy. The Smith-Waterman algorithm’s dynamic programming approach determines the best alignment between two sequences.
We can split the alignment matrix into smaller submatrices and give each submatrix a different GPU thread to parallelize the Smith-Waterman technique.
The alignment scores for the submatrix the thread on the GPU was given control over can then be separately computed once all GPU threads have calculated their alignment scores. Therefore, we can aggregate the data to get the final alignment matrix.
Banded alignment
Using a method known as banded alignment is an additional popular strategy for GPU-accelerated sequence alignment.
The Smith-Waterman algorithm changes by banded alignment. Therefore, it limits consideration to alignments close to one another to minimize the number of comparisons that must be made.
We can use a warp shuffling technique to accomplish banded alignment on a GPU. Warp shuffling allows us to communicate between the different threads in a warp efficiently.
It can transmit the alignment scores for the alignment band’s edges between the various warp threads. The alignment scores for the entire alignment band can thus be efficiently computed in parallel.
GPU-accelerated sequence alignment software
There are several GPU-accelerated sequence alignment software tools available. Some of the most popular tools include:
- CUDA-SW++: CUDA-SW++ is a GPU-accelerated implementation of the Smith-Waterman algorithm. It is one of the most widely used GPU-accelerated sequence alignment tools, and it is known for its speed and accuracy.
- GASAL2: GASAL2 is another GPU-accelerated implementation of the Smith-Waterman algorithm. It is newer than CUDA-SW++, but it is already gaining popularity due to its high performance and ease of use.
- BWA-MEM: BWA-MEM is a popular read mapper that aligns short sequencing reads to reference genomes. It has a GPU-accelerated mode that can significantly speed up the alignment process.
- NovoAlign: NovoAlign is a commercial read mapper with a GPU-accelerated mode. It is known for its high speed and accuracy but is more expensive than other options.
| Tool | Algorithm | Speed | Accuracy | Ease of use | Cost |
|---|---|---|---|---|---|
| CUDA-SW++ | Smith-Waterman | Fast | Accurate | Moderate | Free |
| GASAL2 | Smith-Waterman | Fast | Accurate | Easy | Free |
| BWA-MEM | Burrows-Wheeler transform | Fast | Accurate | Moderate | Free |
| NovoAlign | Burrows-Wheeler transform | Very fast | Very accurate | Easy | Commercial |
Except for NovoAlign, every one of the tools above is free.
The appropriate tool for you will depend on your unique needs and financial situation.
I advise utilizing NovoAlign if you require the quickest and most precise instrument.
But the most expensive tool is NovoAlign. I recommend using CUDA-SW++, GASAL2, or BWA-MEM if your budget is limited. These tools are all rapid and accurate, and they are all free.
GPU-accelerated de novo assembly
Using GPUs to expedite the de novo assembly of genomes and transcriptomes using sequencing data is known as GPU-accelerated de novo assembly.
De novo assembly works well with GPUs since they can handle many computations simultaneously. De novo assembly, which includes comparing numerous sequences to one another, depends on this.
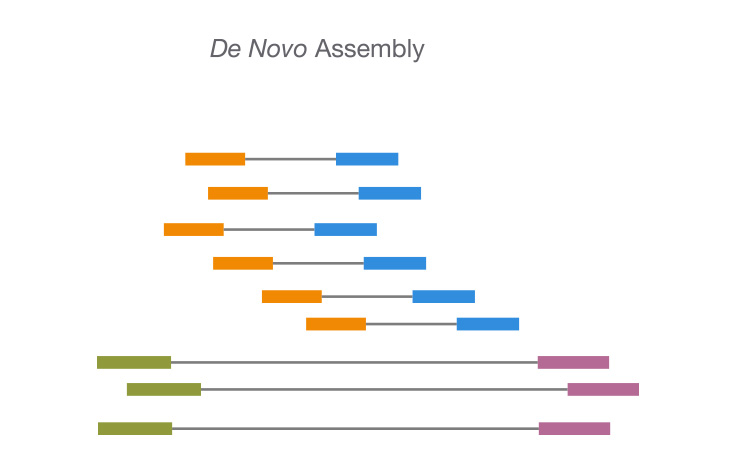
There are numerous different GPU-accelerated de novo assembly algorithms. One such strategy is using a GPU-accelerated variant of the overlap-layout-consensus (OLC) algorithm.
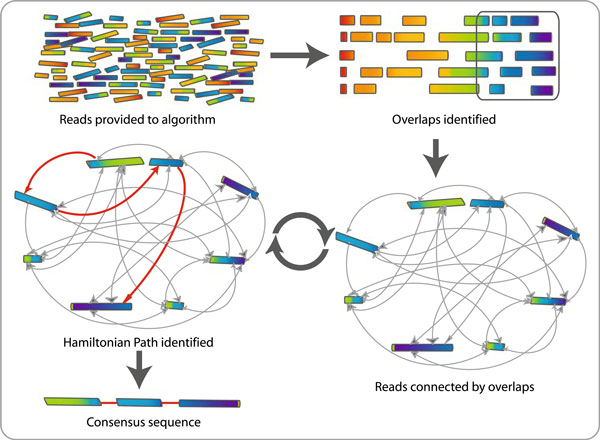
A well-liked de novo assembly approach, the OLC algorithm finds overlaps between sequences and then assembles the overlaps into longer contigs.
A de Bruijn graph is a typical method for GPU-accelerated de novo assembly. A data structure that can be useful to represent the connections between sequences is a de Bruijn graph.
Therefore, De Bruijn graphs can be helpful by GPU-accelerated de novo assembly methods to swiftly and effectively build genomes and transcriptomes.
The de novo assembly process can be considerably sped up using GPU acceleration.
One study, for instance, shows a GPU-accelerated de novo assembler could assemble the human genome in less than two hours.
It can take days or even weeks for conventional CPU-based de novo assemblers to put together a human genome, whereas this is substantially faster.
GPU-accelerated De novo assembly-related software
A de Bruijn graph is first built using SPAdes using the sequencing data. A de Bruijn graph is a type of data structure that shows how overlapping sequences relate. The genome or transcriptome contigs are assembled by SPAdes using the de Bruijn graph.
Contigs are assembled using various algorithms by SPAdes from the de Bruijn graph. Hence, One such strategy is to employ a greedy algorithm.
A greedy algorithm extends contigs until it is unable to develop them further. Using a K-mer method is another typical strategy.
A k-mer technique assembles the contigs from the k-mers after locating all of the k-mers in the de Bruijn graph.
As a de novo genome assembler, Velvet can construct an organism’s genome from brief sequencing data.
Since its initial creation in 2008, it has become one of the most popular de novo assemblers.
A de Bruijn graph must first be built from the sequencing data for Velvet to function.
Although Velvet is a quick and precise de novo assembler, its calculation costs can be high.
This is due to the memory-intensive procedure that Velvet must do to create a de Bruijn graph from the sequencing data.
Velvet, however, can be used on GPUs to quicken the assembly procedure.
A de novo genome assembler, SOAPdenovo. Therefore, it can help assemble genomes of all sizes, from bacteria to people.
Since its initial creation in 2010, it has become one of the most common de novo assemblers.
We can use Both CPUs and GPUs with SOAPdenovo. SOAPdenovo runs much more quickly on GPUs than it does on CPUs.
Because de novo assembly requires comparing numerous sequences to one another, GPUs’ ability to do multiple computations at once is crucial.
MERACulous
De novo metagenomic assembler MERACulous became available for the first time in 2012. It serves to put together genomes from intricate samples of many species.
MERACulous is famous for its capacity to assemble genomes from samples with high levels of microbial diversity and low-quality sequencing data. Therefore, MERACulous constructs genomes from metagenomic data using a variety of methods.
A binning algorithm is one popular strategy. A binning algorithm groups the contigs into bins according to their similarities.
After the contigs have been binned, MERACulous can assemble the genomes from each container using a familiar de novo assembler.
For scientists who need to assemble genomes from large samples or samples with a lot of microbial variety, there are better options than MERACulous.
De novo genome assembler Canu was created for the first time in 2015. It assembles genomes from single-molecule sequencing reads, like those generated by Oxford Nanopore Technologies (ONT) and Pacific Biosciences (PacBio) sequencers.
To begin, Canu creates a de Bruijn graph using the sequencing data. Moreover, Canu builds genomes from single-molecule sequencing readings using various methods.
One such strategy is to employ a greedy algorithm. A greedy algorithm extends contigs until it is unable to develop them further.
Using a k-mer method is another typical strategy. A k-mer technique assembles the contigs from the k-mers after locating all of the k-mers in the de Bruijn graph.
GPU-accelerated taxonomic classification for metagenomic analysis
Classifying bacteria into various taxonomic categories, including species and genera, is taxonomic classification. Therefore, Understanding the diversity and purpose of microbial communities depends on this.
GPUs can speed up taxonomic categorization by making several parallel comparisons between the sequences of unknown microorganisms and reference genomes.
GPU-accelerated taxonomic classification tools for Metagenomics Analysis
- Kraken2
Kraken2 is a fast and precise taxonomic classification tool that uses GPU acceleration. It can classify millions of sequences per second.
Before anything else, Kraken2 builds a database of k-mers or small DNA sequences. Moreover, Kraken2 subsequently determines the taxonomic categories to which the sequencing reads belong by comparing them to the k-mer database.
Kraken2 can classify sequences of all lengths, from small reads to entire genomes. It is beneficial for categorizing massive databases of sequences, such as those produced by metagenomics research.
The initial step in operating a centrifuge is creating a database of k-mers, or short DNA sequences. Centrifuge then determines the taxonomic categories to which the sequencing reads belong by comparing them to the k-mer database.
Several benefits of centrifuge over other taxonomic categorization techniques include:
- It is swift. Centrifuge can classify millions of sequences per second.
- It uses memory quite effectively. Centrifuge can successfully categorize enormous datasets of sequences since it uses less memory while operating.
- Utilization is simple. Therefore, the Centrifuge user interface and documentation are self-explanatory and comprehensive.
GPU-BLAST
The well-known BLAST alignment tool has a GPU-accelerated variant, GPU-BLAST.
It compares them to a reference database of recognized sequences. Therefore, It can be helpful to categorize sequences.
GPU-BLAST is an excellent tool for classifying massive datasets of sequences because it is substantially faster than conventional CPU-based BLAST methods.
It operates by parallelizing the BLAST algorithm on the GPU.
Because BLAST involves comparing numerous sequences to one another, the GPU’s capacity for multitasking makes it perfect for this task.
GPU-accelerated functional annotation
The technique of tagging genes and other genomic characteristics with their known functions is GPU-accelerated functional annotation.
Understanding the biology of an organism and its genes begins with functional annotation.
Therefore, It can be helpful to discover novel genes and comprehend the biological pathways in which genes function. Moreover, it can create brand-new diagnostic and therapeutic tools.
GPU-accelerated functional annotation tools
The software program ANNOVAR annotates genomic variations with known effects. It can annotate variations from many species, such as mice, rats, and humans.
Therefore, because of its reputation for speed and accuracy. ANNOVAR is a prominent tool for annotating variations from genome-wide association studies (GWAS) and other extensive investigations.
For ANNOVAR to function, information regarding known variants must first be retrieved from the database.
The variants with available effects are then annotated by ANNOVAR using this information. ANNOVAR can annotate variants with a wide range of data, such as:
- Gene names
- Transcript IDs
- Exon/intron boundaries
- Protein changes
- Functional predictions
- Regulatory elements
Variant Effect Predictor
A computer program VEP, or Variant Effect Predictor. It helps to forecast how genetic variations may affect how genes function.
It can foretell the outcomes of variations from many species, including mice, rats, and humans.
For VEP to function, data on variants first comes from a database of known variants.
VEP then uses this knowledge to forecast how the variations affect gene function.
The following are some of the factors that VEP can forecast how variations may affect:
- Splicing
- Protein translation
- Protein structure
- Protein function
VEST
The Vienna Suite for Protein Structure Prediction, or VEST, is a set of computer programs that forecasts proteins’ three-dimensional (3D) structure.
For VEST to function, a protein homology model must first be built.
Therefore, A homology model is a 3D protein structure on a previously known protein’s 3D structure.
Using VEST using the homology model, we can improve the 3D structure of the protein. It can use several techniques to improve the protein’s 3D structure.
- Molecular dynamics simulations
- Monte Carlo simulations
- Energy minimization
VEST can predict the Protein complexes’ structures-an interaction between two or more proteins results in the formation of a protein complex.
It employs several techniques to predict the structure of protein complexes, including:
- Docking simulations
- Contact maps
- Distance constraints
Comparison of software for GPU-accelerated functional annotation
| Software | Features | Pros | Cons |
|---|---|---|---|
| ANNOVAR | Supports various annotation databases, including RefSeq, Ensembl, and UniProtKB. Can annotate variants, transcripts, and genomes. | Fast and accurate. User-friendly interface. | It can be computationally expensive for large datasets. |
| VEP | Supports various annotation databases, including Ensembl, RefSeq, and ClinVar. Can annotate variants and transcripts. | Fast and accurate. Supports a variety of annotation formats. | It can be computationally expensive for large datasets. |
| VEST | Predicts the 3D structure of proteins. It can be used to annotate proteins based on their system. | Fast and accurate. Can expect the form of protein complexes. | It can be computationally expensive for large datasets. |
Future advancements in using GPU for Metagenomic Analysis
We use GPUs in several ways, like speeding up metagenomics analysis. But there’s certainly room for improvement in the future. Here are some prospective developments in the use of GPUs for metagenomics analysis in the future:
New algorithms
We can create New algorithms to take advantage of GPUs’ unique qualities and speed up metagenomics analysis.
For instance, fresh techniques can make genome assembly from long-read sequencing data quicker than it is now.
New software tools
We can generate New software tools to make it simpler for researchers to employ GPUs for metagenomics studies.
New software tools could be built to provide graphical user interfaces. Moreover, it automates the process of conducting GPU-accelerated metagenomics analysis pipelines.
More powerful GPUs
As graphics processing units (GPUs) grow more full and affordable, they can speed up metagenomics analysis even more.
For instance, new GPUs could do increasingly tricky metagenomics analysis tasks. It includes the analysis of single-cell metagenomes and the assembly of genomes from even bigger metagenomics datasets.
GPUs to develop new methods for assembling genomes from long-read
Long-read sequencing data is more difficult to compile than short-read sequencing data, but it can reveal more about the structure of genomes.
So, by using GPU, we can create new methods and software tools to swiftly and accurately assemble genomes from long-read sequencing data.
GPUs to develop new methods for analyzing single-cell metagenomes
The study of a single microbe’s genetic makeup is single-cell metagenomics. New software tools and techniques that enable speedy and precise single-cell metagenome analysis may be created using GPUs.
Conclusion
GPUs are a potent instrument that can quicken the processing of several biological research applications, including metagenomics analysis.
While maintaining accuracy and scalability, GPUs can significantly speed up metagenomics analysis.
Assembling genomes from sequencing reads, matching reads to reference genomes.
Moreover, categorizing reads according to taxonomy and functionally annotating reads are just a few ways GPUs are already utilized to speed up metagenomics processing.
Future developments in the use of GPUs for metagenomics analysis are yet possible.
Creating new software tools and algorithms to use GPUs’ unique strengths and speed up metagenomics analysis is possible.
Additionally, GPUs will be able to speed up metagenomics analysis even more as they get cheaper and more powerful.
Overall, the use of GPUs for metagenomics analysis has an auspicious future.
By revolutionizing the analysis of metagenomics data, GPUs have the potential to uncover novel information on the variety and functionality of microorganisms in the environment and the human body.