Abstract
Classical drug design involves a significant investment in time and money for research and development.
Modern computational techniques, such as computational biology, computer-aided clinical medicine, and artificial intelligence, offer the potential to speed up the effectiveness of pharmaceutical research by reducing the effort and expense involved.
As a result of the growing adoption of computational approaches in recent years to improve the quality and safety of drug discovery and pipeline, several novel medications have been certified for commercialization.
The current study applies these fundamental computational approaches to target selection, lead finding, and optimization. Some challenges with using these methodologies for medicine design are also discussed.
We also provide a method for combining several computational approaches with drug discovery and design.
Introduction
Drug development involves several steps, including drug designing, medical trials, and production approval. The process of discovering new drugs is a prolonged, expensive, and difficult one that takes a lot of time and costs ample money.
Target selection, lead discovery, lead optimization, and preclinical screening are all parts of this procedure [1].
Preclinical testing, disease diagnosis, appropriate target identification, efficient molecule identification (including chemical processing and bioactivity screening), and effective target identification are the first steps in the traditional drug discovery process [2].
A developing trend in the pharmaceutical business is high-performance computing (HPC) and artificial intelligence (AI) for drug development and repurposing. By screening massive datasets of chemical compounds, AI may be used to discover possible novel medications.
In contrast, HPC can be used to model how these drugs affect biological systems. The areas of AI and HPC will be covered in this review article, which has the potential to expedite the drug discovery process, lower costs, and enhance patient outcomes.
Computational biology’s contributions to drug development include the following:
- The characterization of ligand-binding molecular processes.
- The recognition of binding/active sites.
- The structure refinement of ligand-target binding poses.
Most of these methods suggest that it’s essential to identify the target protein’s binding and active sites. Certain regions of these binding sites may be used as a guide to modify and improve the original lead molecule and create novel ligand-target protein interactions [3,4,5].
The involvement of the binding site may only sometimes be sufficient to investigate the pathogenic activity. Besides mutations that occur outside the active site, pathosis can also be brought on by structural changes, antibiotic resistance, and expression levels.
Computational biology is a powerful technique for exposing the underlying mechanisms of the target protein and supplying new information, particularly for bio-macromolecular simulation.
Recently, artificial intelligence (AI) has been put out as a viable method for learning from and discovering extensive pharmacological data in drug discovery, which has increased the incidence rates of drug identification.
AI can learn and find new rules for converting enormous datasets from biological research into understandable knowledge [6].
In this article, We have reviewed effective computational methods for drug development and provided an overview of several computational approaches used in drug discovery in this review article. It also covers using VS and other computational tools to expand the chemistry space of new lead compounds and speed the drug development process [7,8].
Role of computational drug discovery and design
Since the 1970s, computational drug discovery and design (CDDD), commonly referred to as computer-assisted drug design, has made considerable advancements. The 3D structures and roles of macromolecules have been identified thanks to the quick progress of both structural and molecular biology [9,10].
In the meantime, as computer science has advanced quickly, graphic workstations have powerful features, high-performance computers have become more common, and so have computational approaches in drug development and molecular simulation.
The production of pharmaceuticals has frequently utilized particle physics, molecular mechanics, molecular dynamics (MD), and combinations of these techniques.
In the past, selling a novel medicine in the pharma industry has been a challenging process that frequently takes more than ten years and requires over one billion dollars.
The effectiveness of drug discovery has been considerably increased by several CDDD techniques that rely on the three-dimensional structures of biological macromolecules (such as proteins and nucleic acids), such as the improved virtual screening methods.
An effective tool in drug research and development, CDDD based on High-performance computer (HPC) combines medicinal chemistry, computational chemistry, and biology via supercomputers.
Recent technologies like grid computing, cloud computing, and accelerating technology like graphics processing units help speed up many CDDD approaches like drug high-throughput screening, MD simulations, and molecular modeling.
Cloud computing, which is often made up of several geographically dispersed supercomputers, is frequently utilized for elevated virtual drug screening to cut down on time and cost.
The total reliance on foreign commercial software has been replaced by the development of numerous novel drug design methods, drug discovery and development tools, and databases featuring the independent intellectual property.
Current approaches for drug development include active compound identification against targets from existing compound libraries, modeling and 3-dimensional predicting of protein folding, and more.
Computational performance has significantly impacted the virtual drug screening model accuracy and enrichment rate. Mass HPC resources are required for cutting-edge drug-related computational genomics, proteomics, and drug design technologies.
Creating the following generation of exascale high-performance computers is necessary for these vital technologies. Exascale computing will be crucial for creating tailored medicines in the next five to ten years [11].
High-throughput screening in drug discovery
The main objective of drug discovery, in most situations, is to find potent target inhibitors through high-throughput screening or virtual screening. The latter uses high-performance computers to choose between cost and precision, whereas the former is more accurate but also more expensive and time-consuming.
Therefore, virtual screening is primarily used over new or unverified targets, mainly in the research labs of academic institutions, to find potent inhibitors and to observe the pathway of these unconfirmed targets.
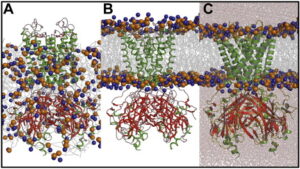
In contrast, high-throughput screening is widely used against well-established targets, primarily in the research labs of the pharmaceutical industry.
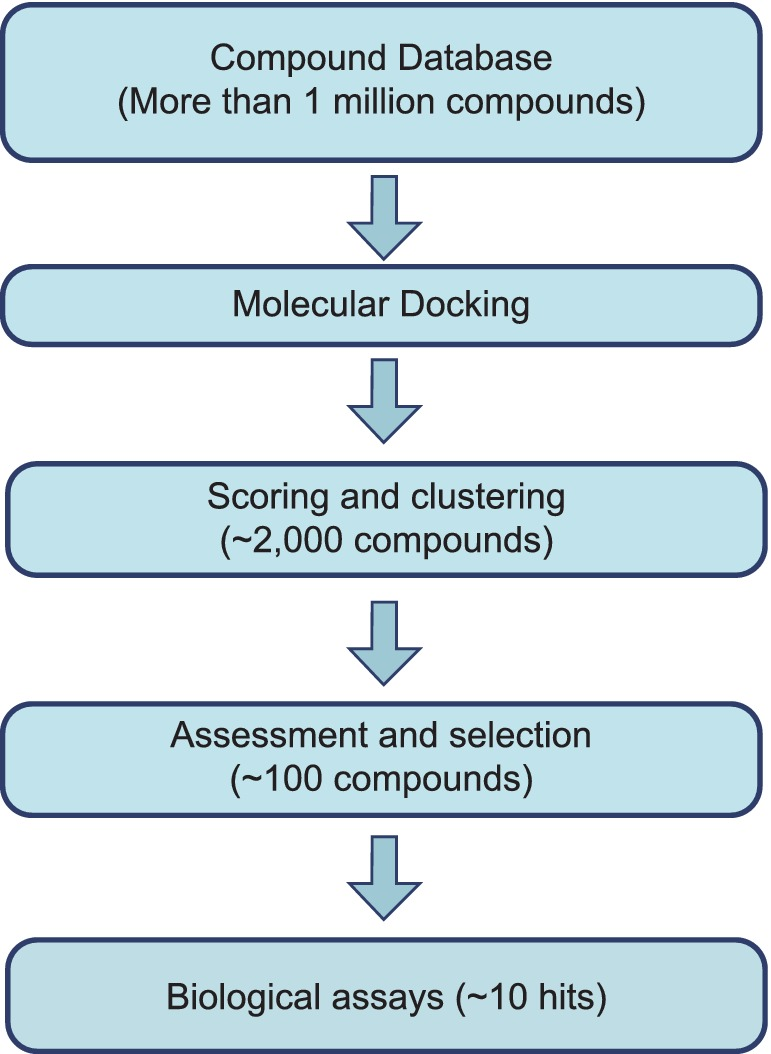
In a typical procedure for virtual screening, a compound database with thousands or even millions of thousands or even millions of entries for small molecules is initially tested using the molecular docking technique against the chosen target protein.
On modern CPUs, molecular docking often takes a little time to minutes while considering the flexibility of tiny molecules and maintaining the protein’s rigidity.
As a result, virtual screening against a target uses a lot of processing power that supercomputers can only offer. The best docking values among thousands of compounds are then preserved for structural grouping.
Drug-like characteristics and molecular docking-generated binding modes serve as the selection criteria. Several techniques are used, such as enzymatic activity trials, enzymatic binding tests, enzymatic selectivity assays, structure-activity relationship investigations, and cell assays, to evaluate the efficacy of these candidate drugs [12].
RNA metabolism, signal transmission, and chromatin remodeling depend on protein arginine methyltransferases (PRMTs). The abnormal activity of PRMTs has been linked to several illnesses, including cancer and cardiovascular conditions.
Virtual screening and radioactive methylation assays were combined to identify numerous hits as PRMT1 inhibitors with micromolar potency. Two of these hits demonstrated even greater potencies than the well-known PRMT inhibitor AMI-1.
Instead of PRMT1, they directly targeted substrate H4 to accomplish inhibition. Additionally, one of these substances significantly reduced the growth of castrate-resistant prostate cancer cells [13].
Target identification using Molecular docking
In addition to being used in virtual drug discovery screening, molecular docking, the core tool of computer-aided drug design, also helps target identification.
The pleiotropic lipid mediator sphingosine-1-phosphate (S1P) can function independently of its membrane receptors through an internal mechanism.
One of the isoenzymes that produce S1P is sphingosine kinase 2 (SPHK2), which is connected to histone H3 and controls histone acetylation by binding to the histone deacetylases HDAC1 and HDAC2 and inhibiting their enzymatic activity.
In the nucleus, SPHK2 phosphorylates FTY720, which then binds to and inhibits HDACs to promote specific histone acetylation.
Contrarily, FTY720 (fingolimod), an FDA-approved medication for the therapy of multiple sclerosis, has beneficial impacts on the central nervous system, which are separate from its effects on immune cell trafficking; however, the mechanism is not yet fully understood.
After molecular docking revealed the possible binding manner of FTY720 inside the pocket of HDAC, it was determined that FTY720 might be an effective adjuvant therapy to speed up the extinction of unpleasant memories by targeting HDACs [14,15].
Target prediction for drug repositioning using CDDD
Drug targets are frequently linked to a desired therapeutic outcome. Identifying potential interactions between ligands and proteins aids in forecasting the therapeutic effects or adverse effects of drug-like compounds.
The identification and prediction of targets are crucial for drug discovery and research. However, it is time-consuming and challenging if done solely by experimental methods. Therefore, it is critical to creating effective computational techniques that can identify the targets of chemical molecules.
Recently, Li et al. created the reverse ligand-protein docking tool TarFisDock to explore the possible therapeutic target database for probable ligand-protein interactions.
A successful application of TarFisDock is considered the discovery of the enzyme peptide deformylase (the target of the natural product N-trans-caffeoyltyramine) in Helicobacter. To develop a web server named PharmMapper and anticipate prospective drug targets, Liu et al. suggested a reverse pharmacophore mapping technique.
Due to the exponential rise of bioactivity data, it has become increasingly challenging to handle the large-scale interaction data of small compounds and their targets using conventional computational approaches.
Therefore, target prediction is a massive data issue. Liu et al. developed a ligand-based target fishing technique based on data fusion and 2D fingerprint similarity rankings. This approach is suitable for large-scale drug target determination due to its simple algorithm and short calculation time [16,17].
A user-friendly website called TarPred was created to make this technology more practical by predicting query compounds’ therapeutic advantages and negative consequences. You can obtain <a